2Department of Biochemistry and Molecular Biology, Dhaka Shishu Hospital, Dhaka, Bangladesh
3Department of Paediatric Haematology Thalassaemia and Oncology, Dhaka Shishu Hospital, Dhaka, Bangladesh
![]() 10.3329/bmrcb.v44i2.38688
10.3329/bmrcb.v44i2.38688 ![]() http://orcid.org
http://orcid.org
A total of 100 Bangladeshi beta thalassaemia carrier subjects were analysed by allele specific primers using Amplification Refractory Mutation System – Polymerase Chain Reaction. Among these, four common mutations were found in 90 cases (90.0%), five less common mutations in 9 cases (9.0%) and a rare mutation in 1 case (1.0%). Among the four common mutations, IVS 1-5 (G-C) was the most common beta thalassaemia mutation and found in 63.0% cases, followed by Cd 30 (G-C) in 18.0%, Fr 8/9 (+G) in 5.0% and Fr 41/42 (-TTCT) in 4.0% respectively. Among the five less common mutations, Cd 16 (-C) was found in 3.0%, -90 (C-T) and IVS 1-130 (G-C) were seen in 2.0% each and remaining Cd 15 (-T) and Cd 15 (G-A) were detected in 1.0% each. The rare mutation was -29 (A-G), observed in one case (1.0%). With the application of this knowledge, it will help us for prenatal diagnosis and genetic counselling in Bangladesh for prevention of the disease.
Keywords: Beta thalassaemia carrier, ARMS-PCR, Bangladeshi mutation, Prenatal diagnosisThalassaemia is a common and debilitating autosomal recessive haemoglobin disorder affecting many populations in the world including Bangladesh. In Bangladesh, two types of thalassaemia are common. One of them is beta thalassaemia major and the other one is HbE beta thalassaemia, with overall prevalence of beta thalassaemia trait 4.1% and HbE trait 6.1%. It is estimated that more than seven thousand children are born with thalassaemia each year in the country.1 Thalassaemia causes severe blood dependent anaemia and to correct the anaemia, blood transfusion is given every month or more frequently. Frequent blood transfusion causes deposition of iron in the body which is toxic and causes damage to organs like liver, heart, pancreas leading to their failure. Drugs used to remove iron are expensive. Majority of the thalassaemic patients cannot afford adequate treatment and die at an early age.
Prevention of births of thalassaemic children is cost effective.2 Carrier detection, genetic counselling and prenatal diagnosis are important for prevention of the disease. Among these, prenatal diagnosis has been done all over the world where the problem of thalassaemia exists like Pakistan, India, Thailand, Iran, Cyprus and Turkey.3-10 Cyprus had one of the highest carrier rates in the world, with an estimated carrier rate of one in seven but now there is not a single birth of thalassaemic child since last five years with the start of prenatal diagnosis and abortion of affected child with the parent’s consent.9 There has also been significant fall of births of thalassaemic children in Iran after the introduction of prenatal diagnosis and abortion made legal.8
Before introducing prenatal diagnosis in Bangladesh, it is important to know the mutation pattern of beta thalassaemia as mutations in the beta globin chain is the common cause of thalassaemia in the country. Alpha thalassaemia has rarely been reported. There are more than two hundred types of beta thalassaemia mutations that cause this disease.11 Fortunately, in every country, there are a few mutations of beta-globin genes. In Indian sub-continent, several mutations are detected, among which five mutations are selected for our study which accounted for 84.50% of total mutations.12 Mutation pattern of Indian sub-continent has been selected because no relevant studies have been done on mutation pattern of beta thalassaemia in Bangladesh.
The objective of the study was to assess the mutation pattern of beta thalassaemia in Bangladeshi population as the application of this knowledge would facilitate genetic counselling and prenatal diagnosis.
A cross sectional study was carried out in the Department of Biochemistry and Molecular Biology at Dhaka Shishu (Children) Hospital, Dhaka, Bangladesh during August 2014 to October 2014. A total of 100 blood samples were collected from beta thalassaemia carrier subjects. All the traits were parents of known beta thalassaemia major, who came to Dhaka Shishu Hospital Thalassaemia Centre for blood transfusion of their children. For further confirmation of beta thalassaemia trait, complete blood count (CBC) was done in all cases by automated haematology analyzer (Mythic-18, France) to evaluate the values of red blood cell (RBC) parameters and carrier status was confirmed by High Performance Liquid Chromatography (HPLC) (D-10TM Dual Program, Bio-Rad Laboratories, Hercules, CA, United States). The value of HbA2 level >3.5% was diagnosed as beta thalassaemia trait and included in the study for mutational analysis.11,13
Two millilitre of blood was taken in EDTA (ethylene diamine tetra-acetic acid) vial from each case for CBC and HPLC and additional 2 ml of blood was taken in another EDTA vial for DNA analysis after taking verbal consent. DNA extraction was done in all 100 samples by Genomic DNA Mini Kit blood protocol, provided by Geneaid.14 Then mutation in the beta globin chain was analysed by allele specific primers using Amplification Refractory Mutation System - Polymerase Chain Reaction (ARMS-PCR).15
Initially, DNA analysis was done with five mutations present in Indian sub-continent which accounted for 84.50%.12 These mutations were IVS 1-5 (G-C), Cd 30 (G-C), Fr 8-9 (+G), Fr 41-42 (-TTCT) and 619 bp deletion. Out of 5 mutations, 4 mutations were detected in this study in 90 cases out of 100 samples. The mutation 619 bp deletion was not identified.
Of the remaining 10 cases, DNA analysis was done with 7 less common mutations also found in Indian sub-continent which were Cd 16 (-C), IVS 1-130 (G-C), IVS 1-130 (G-A), -90 (C-T), Cd 15 (G-A), Cd 15 (-T) and Cd 30 (G-A).16 Among these, 5 less common mutations i.e. Cd 16 (-C), IVS 1-130 (G-C), -90 (C-T), Cd 15 (G-A) and Cd 15 (-T) were detected in 9 cases out of 10 samples.
To find out the mutation of the last one case, DNA sequencing (using 310 Genetic Analyzer, Applied Biosystem, USA) was done where -29 (A-G), a rare mutation was detected.
DNA analysis was done in 100 carriers of beta thalassaemia who came to Dhaka Shishu Hospital Thalassaemia Centre for blood transfusion of their children. Among these, 65 were female and 35 were male between the ages of 17 and 55 years with mean age of 29.48±7.33 years. The females over number the males because most of the thalassaemic children came to the hospital accompanied by their mothers.
The values of RBC parameters and HbA2 level of 100 samples of beta thalassaemia traits were computed and analysed by SPSS Version 18 to find out the maximum, minimum and mean values of Red Blood Cell count (RBC count), Haemoglobin (Hb) level, Mean Cell Volume (MCV), Mean Cell Haemoglobin (MCH), Mean Cell Haemoglobin Concentration (MCHC), Red Blood Cell Distribution Width (RDW) and HbA2 level. These values were compared with normal adult values.17
Parameters |
Beta thalassaemia trait |
Normal adult values17 |
|||
|---|---|---|---|---|---|
Maximum |
Minimum |
Mean ± SD |
|||
RBC Count (x 1012/L) |
Men |
7.19 |
4.15 |
5.90 ± 0.62 |
5.0 ± 0.5 |
Women |
6.75 |
4.35 |
5.33 ± 0.48 |
4.3 ± 0.5 |
|
Hb level (gm/dl) |
Men |
14.40 |
9.30 |
11.80 ± 1.16 |
15 ± 2.0 |
Women |
12.20 |
9.20 |
10.44 ± 0.75 |
13.5 ± 1.5 |
|
MCV (fl) |
76 |
51 |
60.17 ± 3.51 |
92 ± 9 |
|
MCH (pg) |
30 |
16 |
19.66 ± 1.84 |
29.5 ± 2.5 |
|
MCHC (gm/dl) |
36 |
30 |
32.56 ± 0.94 |
33 ± 1.5 |
|
RDW (%) |
19 |
12 |
14.83 ± 1.33 |
12.8 ± 1.2 |
|
HbA2 (%) |
7.20 |
3.70 |
5.60 ± 0.68 |
2.2 – 3.5 |
|
Among the 100 samples for analysis of mutations, 4 mutations were found in 90 cases (90.0%) which can be considered as common mutations in our study, less common mutations were 5, seen in 9 cases (9.0%) and rare mutation found in one case (1.0%).
Among the 4 common mutations (90.0%), IVS 1-5 (G-C) was the most common mutation identified in 63 cases with the frequency of 63.0%. The second common mutation was Cd 30 (G-C), found in 18 cases (18.0%). The frequency of other two common mutations, namely Fr 8/9 (+G) and Fr 41/42 (-TTCT) were found in 5 cases (5.0%) and 4 cases (4.0%) respectively. (table II and figures 1, 2, 3 and 4)
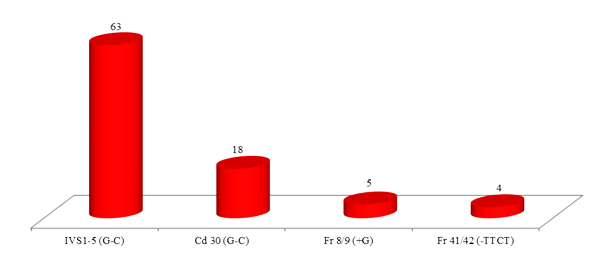
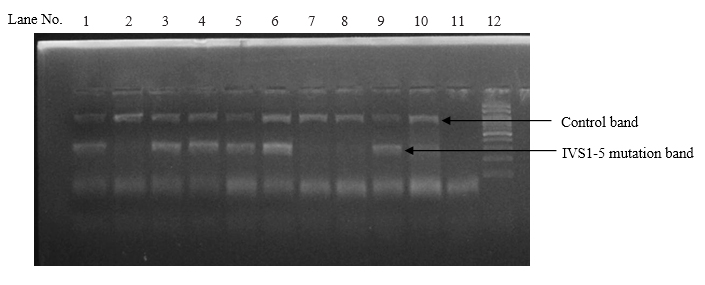
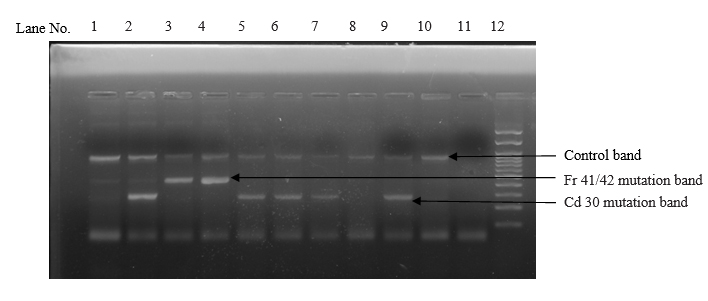
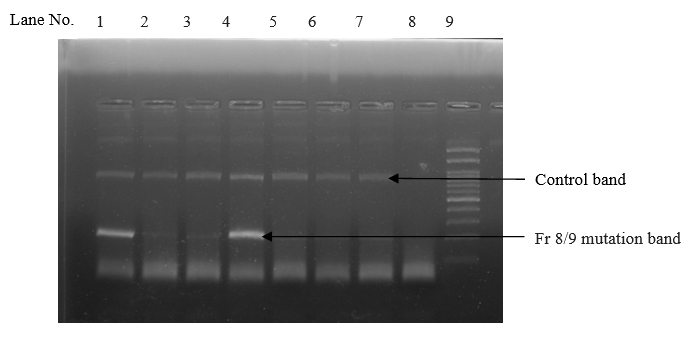
Among the 5 less common mutations (9.00%), Cd 16 (-C) was found in 3 cases (3.00%), IVS 1-130 (G-C) in 2 cases (2.0%), -90(C-T) in 2 cases (2.00%), Cd 15 (-T) in one case (1.0%) and Cd 15 (G-A) in one case (1.0%) respectively. (table II)
The mutation of remaining one case (1.0%) was -29 (A-G), a rare mutation detected by DNA sequencing.
Sl. no. |
Mutation |
Frequency (%) |
Number of beta trait |
|---|---|---|---|
1. |
IVS1-5 (G-C) |
63.00 |
63 |
2. |
Codon 30 (G-C) |
18.00 |
18 |
3. |
Fr 8/9 (+G) |
5.00 |
05 |
4. |
Fr 41/42 (-TTCT) |
4.00 |
04 |
5. |
Cd16 (-C) |
3.00 |
03 |
6. |
-90 (C-T) |
2.00 |
02 |
7. |
IVS1-130 (G-C) |
2.00 |
02 |
8. |
Cd 15 (-T) |
1.00 |
01 |
9. |
Cd 15 (G-A) |
1.00 |
01 |
10. |
-29 (A-G) |
1.00 |
01 |
Total |
100.00 |
100 |
|
A total of 100 beta thalassaemia carrier subjects were screened by allele specific primers using ARMS-PCR method for detection of mutation pattern of beta thalassaemia in Bangladeshi population. Among the 100 cases, the most common beta thalassaemia mutation was IVS 1-5 (G-C), found in 63 cases (63.0%) followed by Cd 30 (G-C) in 18 cases, Fr 8/9 (+G) in 5 cases and Fr 41/42 (-TTCT) in 4 cases with frequencies of 18.0%, 5.0% and 4.0 % respectively. These 4 mutations were accounted for 90.0% of the total and can be considered as common mutations in our country. The five less common mutations were Cd 16 (-C) in 3 cases (3.0%), IVS 1-130 (G-C) and -90 (C-T) found in 2 cases (2.0%) each and the remaining less common mutations were Cd 15 (G-A) and Cd 15 (-T) seen in one case (1.0%) each. The rare mutation was -29 (G-A) detected in one case (1.0%).
Among the 4 common mutations, IVS 1-5 (G-C) was the most common mutation in our study, being seen in 63.0%, which was also most common mutation with varied frequencies as reported by others.16,18-24
The second common mutation was Codon 30 (G-C) with a frequency of 18.0%. This mutation was also seen by other authors but with low frequencies.16,18, 22-25
The third common detected mutation was Fr 8/9 (+G), accounted for 5.0%. Nearly similar low observation was also seen by some authors21-23 but in contrast to others,19,26-27 where percentage of Fr 8/9 (+G) was reported higher. The observed fourth common mutation was Fr 41/42 (-TTCT), representing 4.0% of the total. Similar low frequency was reported by some, but higher percentage reported by others.16,19,21-23,25-27
Among the 5 less common mutations, this study showed Cd 16 (- C) mutation was 3.0% and also observed by other workers with low frequencies.16,20,22,23,27 -90 (C-T) mutation was accounted for 2.00% and this is consistent with the findings seen by other authors.23,24 The present study detected IVS 1-130 (G-C) mutation was 2.0% which was also seen by other authors with low frequencies.22,23 The remaining less common mutations were Cd 15 (G-A) and Cd 15 (-T) showing 1% each. Cd 15 (G-A) mutation was seen by several authors with varied frequencies16,18,21,23,25,27 and Cd 15 (-T) observed by others with low frequencies.22,23
One (1.0%) rare mutation was detected in our study which was -29 (A-G) and this mutation was also detected in India but not in Pakistan and Sri Lanka with low frequency.16
By comparing the mutations of the present study with other recent studies of Bangladesh showing that there were significant variations of mutations found in beta thalassaemia trait of Bangladeshi population. These mutations were 619 bp deletion, CD1 (T-A), CD2 (C-A), CD2 (T-C), IVSII-16 (G-C), IVSII-81 (C-T), -80 (T-C), 92 (G-C) and 92 (C-G) but not detected in the present study. All of these mutations were reported as lower frequencies except CD2 (T-C) and IVSII-81 (C-T) where reported as higher frequencies.23,24,28 Studies with larger population are needed to find out these mutations which may help proper genetic counselling as well as control of this disorder in Bangladesh.
Regarding RBC parameters, beta thalassaemia traits in general have reduced MCV and MCH with marginally reduced or normal MCHC. RBC count is often normal even mild anaemia or may be increased. Hb level is usually normal or mildly reduced.29 The present study also showed the similar findings. The value of RDW is usually normal in beta thalassaemia trait and increased in iron deficiency anaemia.30 This study showed both normal and increased RDW in beta thalassaemia traits suggesting that those traits revealed increased RDW may be associated with iron deficiency anaemia which could not be confirmed by serum iron profile estimation due to financial constraint.
Conclusion
Mutation pattern of beta thalassaemia carrier subjects were analysed and four mutations were found common in Bangladeshi population which accounted for 90.0%. With the application of this knowledge, it will help to start prenatal diagnosis and genetic counselling in the country for prevention of thalassaemia.
Acknowledgment
The authors gratefully acknowledge the BMRC for financial support to conduct this study. We are also thankful to all beta thalassaemia carrier subjects who provided blood samples to carry out the study. The co-operation and technical support of staff of the Pathology Department are also appreciated.
References
- Khan WA, Banu B, Amin SK, Selimuzzaman M, Rahman M, Hossain B et al. Prevalence of Beta thalassaemia trait and Hb E trait in Bangladeshi school children and health burden of thalassaemia in our population. DS (Child) HJ. 2005;21:1-6.
- Leung KY, Lee CP, Tang MHY, Lau ET, Ng LKL, Lee YP et al. Cost effectiveness of prenatal screening for thalassaemia in Hong Kong. Prenat Diagn. 2004;24:899-907.
- Ahmed S. Prenatal diagnosis of β-thalassaemia: 12 year’s experience at a single laboratory in Pakistan. Prenat Diagn. 2007;27:1224-27.
- Tamhankar PM, Agarwal S, Arya V, Kumar R, Gupta UR, Agarwal SS. Prevention of homozygous beta thalassemia by premarital screening and prenatal diagnosis in India. Prenat Diagn. 2009;29:83-88.
- Thakur (Mahadik) C, Vaz F, Banerjee M, Kapadia C, Natrajan PG, Yagnik H et al. Prenatal diagnosis of beta-thalassaemia and other haemoglobinopathies in India. Prenat Diagn. 2000; 20:194-201.
- Fucharoen S, Winichagoon P, Thonglairoam V, Siriboon W, Siritanaratkul N, Kanokpongsakdis S et al. Prenatal diagnosis of thalassaemia and hemoglobinopathies in Thailand: experience from 100 pregnancies. Southeast Asian J Trop Med Public Health. 1991;22:16-29.
- Khorasani G, Kosaryan M, Vahidshahi K, Shakeri S, Nasehi MM. Results of the National Program for prevention of beta thalassemia major in the Iranian province of Mazandaran. Haemoglobin. 2008;32:263-71.
- Joulaei H, Shahbazi M, Nazemzadegan B, Rastgar M, Hadibarhaghtalab M, Heydari M et al. The diminishing trend of beta thalassaemia in Southern Iran from 1997 to 2011: The impact of preventive strategies. Haemoglobin. 2014;38:19-23.
- Bozkrut G. Results from the North Cyprus Thalassaemia Prevention Program. Haemoglobin. 2007;31:257-64.
- Mendilcioglu I, Yakut S, Keser I, Simsek M, Yesilipek A, Bagci G. Prenatal diagnosis of thalassemia and other hemoglobinopathies in Southwestern Turkey. Haemoglobin. 2011;35:47-55.
- Renzo G. Screening and diagnosis for haemoglobin disorders. In: Prevention of thalassaemias and other haemoglobin disorders, vol.1. Renzo G, Eleftheriou A, Synodinos JT, Old J, Petrou M, Angastiniotis M (eds). Nicosia, Cyprus: Thalassaemia International Federation Publication; 2003. p. 34-60.
- Varawalla NY, Old JM, Sarkar R, Venkatesan A, Weatherall DJ. The spectrum of β- thalassaemia mutations on the Indian subcontinent: The basis for prenatal diagnosis. Br J Haematol. 1991;78:242-47.
- Hoffbrand AV, Moss PAH. Genetic disorders of haemoglobin. In: Essential haematology. Hoffbrand AV, Moss PAH (eds). 6th ed. West Sussex, UK: Wiley- Blackwell Publishing; 2011. p. 88-107.
- Vogelstein B, Gillespie D. Genomic DNA Mini Kit (blood/cultured cell) protocol Proc Nati Acad Sci. (1979) USA 76.
- Newton CR, Graham A, Heptinstall LE, Powell SJ, Summers C, Kalsheker N et al. Analysis of any point mutations in DNA. The amplification refractory mutation system (ARMS). Nucl Acids Res 1989;17:2503-16.
- Black ML, Sinha S, Agarwal S, Colah R, Das R, Bellgard M et al. A descriptive profile of β- thalassaemia mutations in India, Pakistan and Sri Lanka. J community Genet. 2010;1:149-57.
- Bates I, Lewis SM. Reference ranges and normal values. In: Dacie and Lewis Practical Haematology. Bain JB, Bates I, Laffan MA, Lewis SM (eds). 11th ed. UK: Elsevier Churchill Livingstone Publishing; 2011. p 11-22.
- Bandyopadhyay A, Bandyopadhyay S, Chowdhury MD, Dasgupta UB. Major beta globin gene mutation in Eastern India and their associated haplotypes. Hum Hered. 1999;49:232-35.
- Cristopher AF, Kumari A, Chaudhary S, Hora S, Ali Z, Agrawal SC. Unique pattern of mutations in β-Thalassemia patients in Western Uttar Pradesh. Indian Hum Genet. 2013;19:207-12.
- Gupta A, Sarwai S, Pathak N, Agarwal S. Beta-globin gene mutations in India and their linkage to β-haplotypes. Int J Hum Genet. 2008;8:237-41.
- Satpute SB, Bankar MP, Momin AA. The prevalence of β-thalassemia mutations in South Western Maharashtra. Indian J Clin Biochem. 2012;27:389-93.
- Fisher CA, Premawardhena A, Silva SD, Perera G, Rajapaksa S, Olivieri NA et al. The molecular basis for the thalassaemia in Sri Lanka. Br J Haematol. 2003;121:662-71.
- Chatterjee T, Chakravarty A, Chakravarty S, Chowdhury MA, Sultana R. Mutation Spectrum of β-Thalassemia and Other Hemoglobinopathies in Chittagong, Southeast Bangladesh. Hemoglobin. 2015; 39: 389-92.
- Sultana GNN, Begum R, Akhter H, Shamim Z, Rahim MA, Chaubey G. The Complete Spectrum of Beta (β) Thalassemia Mutations in Bangladeshi Population. Austin Biomark Diagn. 2016; 3:1024
- Kukreti R, Dash D, Vineetha KE, Chakravarty S, Das SK, De M et al. Spectrum of thalassemia mutations and their association with allelic sequence polymorphisms at the globin gene cluster in an Eastern Indian population. Am J Hematol. 2002;70:269-77.
- Khan SN, Riazuddin S. Molecular characterization of β-thlassaemia in Pakistan. Haemoglobin. 1998;22:333-45.
- Khattak SAK, Ahmed S, Anwar J, Ali N, Shaikh KH. Prevalence of various mutations in beta thalassaemia and its association with haematological parameters. J Pak Med Assoc. 2012;62:40-43.
- Ibn Ayub M, Moosa MM, Sarwardi G, Khan W, Khan H, Yeasmin S. Mutation analysis of the HBB gene in selected Bangladeshi beta-thalassemic individuals: presence of rare mutations. Genet Test Mol Biomarkers. 2010; 14:299-302.
- Firkin F, Chesterman C, Penington D, RushB. Disorder of Haemoglobin Structures and Synthesis. In: de Gruchy's Clinical Haematology in Medical Practice. Firkin F, Chesterman C, Penington D, Rush B (eds). 5th ed. Carlton Victoria, Australia: Blackwell Science Ltd; 1989. p 137-71.
- Briggs C, Bain BJ. Basic haematological techniques. In: Dacie and Lewis Practical Haematology. Bain JB, Bates I, Laffan MA, Lewis SM (eds). 11th ed. UK: Elsevier Churchill Livingstone Publishing; 2011. p 23-56.